I’ve seen a lot of talk recently on the possible roles of AI in clinical research. I say “roles” plural, because really it’s not a simple case of whether or not AI will enter the clinical research arena (it already has to some extent), but really how best can AI be utilized to actually improve the process.
In theory, an AI could be used in many of the roles that are currently human. With certain limitations and caveats, AI has been shown to be fast and effective at producing written content or organizing data – both things that are vitally important in the design and reporting of clinical trials. AI models can also be trained to identify and track signals in safety data or find patterns in protocol deviations that might otherwise be too subtle to see. An AI might find an efficient CRA scheduling pattern for site visits to maximize on-site time and minimize travel, and a predictive model based on social media posts and web searches might predict the next location for an infectious disease outbreak so that local study sites can be brought online quickly.
But those limitations and caveats I mentioned above are the weak link in all things AI-related, and I do not for second think that we’re at the point where AI can be considered anything other than yet another tool to draw upon when performing clinical research.
Google’s experiment with using Reddit as a data source for training its Gemini AI is a lesson in the absurdity of allowing an AI model to teach itself. Without human oversight, and lacking any kind of “common sense” to filter out clearly ridiculous text, the kind of answers that were generated to fairly mundane questions ranged from amusing to dangerous. In our context, using AI to perhaps assist in generating protocols, informed consent documents, or any kind of literature, by training it on a database of existing examples, would potentially have the same risks. Aside from the fact that the protocols are proprietary information for each sponsor, protocols are notorious for requiring amendments to fix errors or provide clarifications. They are inherently imperfect, as much as we all try to get them perfect! Even if the protocol doesn’t have errors as-written, there are often compromises based on limitations of time, pre-existing data, drug tolerability, subject preferences, or any of a myriad of reasons to choose a particular timeline or set of measurable outcomes. In fact, as research should build upon what has gone before, it makes no sense to simply use old examples in the hope of drafting a novel approach that might be easier, faster, or cheaper to execute.
My experiences in AI training over the last year have been eye-opening, because of the sheer number of people and hours required to properly try to craft and steer an AI towards even a semblance of mediocrity. It was amusing because there the “contamination” of data was at times in both directions – as an excess of incorrectly formatted math problems were offered up an examples, the model started producing solutions written using the same incorrect format, and as the humans were increasingly exposed to AI-generated content, their own human content began to look more and more AI-generated, such that it began to get flagged as such and rejected!
With regards to clinical trials in particular, one area that I found concerning was with medical knowledge and reasoning. It was very obvious that very few actual medical experts were contributing to the process, as it would have been prohibitively expensive to pay for them. With limited knowledge and experience, training examples were overly simplistic and incorrect, and often relied on basic pattern recognition without a logical understanding of why something is what it is. Historically, AI models have been very good at pretending to be smart, while actually falling way down on standard IQ assessments. If the people being used to guide and train the factual knowledge and logical reasoning of AI in these specialized areas aren’t themselves highly knowledgeable and intelligent, then the model truly is doomed. After all, we’ve already established that they do a very poor job of training themselves!
My point here is that, even if we are able to fund and task the experts to help train up AI models on the kind of specialized work required in clinical trials, it’s still going to require (knowledgeable, experienced, intelligent) human oversight to “check their work”. As such, AI might indeed save us a ton of time and typing, but I don’t think it’s going to be the case of asking “Hey Siri, draft me a phase 1 dose escalation study to find the maximum tolerated dose of generimab” and simply emailing it off to the FDA. There is a very real risk of the proponents of AI over-promising and under-delivering. In the specific area of medical monitoring and subject safety, I would not trust an AI to be able to apply the same clinical reasoning skills of a trained physician, no matter how capable they might appear in offering up diagnoses or treatment plans (to be fair, most AI training steers specifically away from this – but my concerns stem from specific attempts to address this niche). Although I have previously argued that safety monitoring is surprisingly specialty-agnostic, compared to protocol design and study start-up activities, one thing we all have in common is having had the same kind of experience-based training. We’re also at the end of the bell curve that has typically been separate from the AI models, where being able to logically reason and apply our knowledge is far more important than simply knowing facts.
Lastly, and this is a huge concern, is the phenomenon of “hallucination”. This is the situation where an AI model, faced with an inability to answer a question due to a lack of factual knowledge or an inability to logically reason its way through a problem, simply makes something up. Even when explicitly instructed NOT to do this, and to instead say something like “I’m sorry, I’m afraid I can’t do that…”, AI seems to be inherently a people-pleaser. This could result in false statements in investigator brochures or consent forms, in inaccurate study timelines as enrollment rates are hallucinated based on non-existent data, or in inaccurate medical coding or protocol deviations. AI-generated hallucinations are by their very nature IMPOSSIBLE to detect from an accurate outcome, unless you know more and better than the AI. And if that is the case, then why are you using the AI to do the work in the first place…?
With all of that said, do I think that we’ll see more AI in clinical research? Yes, I do. Most importantly, AI is quite simply getting better. Recently, and for the first time, an AI model demonstrated an IQ that was above 100 – suggesting some truly useful logical reasoning is being performed. As an industry, AI software companies can learn from the errors and hopefully train up their models on legitimate content with legitimate guidance, and if the tasks are highly specialized and limited then the advantage of an accurate, fast, tireless entity is obvious. Even in areas where human oversight is expected and required, there’s nothing to stop AI from being a useful screening tool and an assistant to tasks such as medical monitoring or safety signal detection.
I am leery of putting too much stock into the promises of AI just yet, but clearly several groups are working on trying to provide meaningful solutions to the clinical research world and things are likely to improve. One thing is for sure though, they won’t improve unless people are willing and able to put the work into refining and optimizing the AI models for these purposes. If you get the opportunity to contribute to this work in any way, I say go for it!
It’s taken me a little while to put virtual pen to paper in response to the appalling report from Reuters that revealed how an official US-government-sanctioned campaign set out to deliberately undermine COVID vaccination efforts in the Philippines.
Through phony internet accounts meant to impersonate Filipinos, the military’s propaganda efforts morphed into an anti-vax campaign. Social media posts decried the quality of face masks, test kits and the first vaccine that would become available in the Philippines – China’s Sinovac inoculation.
Reuters
The program, which apparently ran from 2020 through the end of the Trump administration and months into 2021, was a deliberate act of propaganda to undermine the influence of China on the global COVID vaccine stage (which the USA was not contributing to at all at the time). While Americans were “not targeted” by the misinformation, that specifically set out to disparage and undermine the Sinovac vaccine that was in widespread use in that region at the time, there cannot be any doubt that the lives of locals were placed at risk. As an infectious disease specialist who has dealt with antivaccine misinformation for all of their professional career, this is simply unacceptable to me. Regardless of the relative merits of one vaccine over another, or the petty machinations of the political regimes, during a global pandemic it is an abject failure of humanity to deliberately place other human beings in harm’s way.
This isn’t the first time that vaccines have been used as tactical tools – in the hunt for Bin Laden the CIA used a hepatitis vaccine program to collect DNA from children in an attempt to discover links to the terrorist in hiding. When the ruse became public, vaccine workers (even those with nothing to do with the program) were targeted and the polio eradication campaign stalled, with effects that have taken over a decade to even start to undo.
In response to the Pentagon’s efforts to undermine China, Filipino vaccine rates were woefully low, to the extent that the Government had to threaten jail for those who refused vaccination. When the US was approaching herd immunity at 65% coverage, in June 2021 the Philippines had only about 2% of their population immunized.
It takes a special kind of heartless disregard for science, and the welfare of other human beings, to promote this kind of act and go along with it. I suppose should we expect anything less, coming from a political leader who downplayed the seriousness and very existence of the problem and then politicized masking and social distancing, even as they sent protective equipment to their political allies. After vaccines became widely available, the anti-science mindset among Republicans was responsible for a 43% higher rate of excess deaths compared to Democrats in Florida and Ohio.
Arguably, deliberately undermining vaccine programs for political gain is tantamount to poisoning a water supply, which is literally a war crime. I cannot overstate how upset I am about this entire story – we as the experts have a hard enough time educating a scared and skeptical public about the measures that we are recommending to literally save them from themselves, without having our work attacked and undone by the powers that be.
One of the first things I tell someone when they ask me about doing clinical research is to make sure they have a good study team, and a good study team starts with a good study coordinator.
The role of a study coordinator (also called a clinical research coordinator or CRC) can be highly varied or very specialized, depending on the site and how they have their research organized. I have worked in both settings, and in fact I cut my clinical trial teeth 20 years ago in the role of a CRC where I was responsible for basically every task in the execution of the clinical trial. Think I’m kidding? Here’s a list off the top of my head…
Re-wrote the industry protocol for IRB submission.
Drafted/edited ICF for IRB submission.
Completed the IRB submission, answered IRB questions.
Created all source documents for the visits.
Pre-screened patients.
Met patients in clinic, consented, screened.
Ensured study drug/vaccine orders were placed properly.
Collected blood and urine specimens.
Processed blood and urine specimens (centrifuge, aliquoting).
Packaged and shipped specimens.
Met subjects for follow-up visits in clinic.
Dealt with queries from the clinical monitor.
Made calls to the medical monitor.
Completed CRFs.
Met with clinical monitor for monitoring visits.
Called subjects for telephone follow-up, tracked subject schedules.
Called external offices for medical records.
Completed/communicated SAE reports.
Trained other study staff (and non-study staff) on the clinical trial.
Now, not every study coordinator can do all of those tasks, but as a Principal Investigator a lot of those are practically impossible tasks to fit into their existing schedule. For this reason a study team often consists of people with diverse skills and roles – for example a study nurse for procedures and assessments, and a data manager for CRF data entry and query responses. It should be clear though that a lot of the front-line work that’s required for running a study site requires having a reliable and smart person on the study team as the study-coordinator. Because clinical research is so tightly regulated, you can’t afford to have someone who isn’t careful and thorough on the team. You can also see the advantages of having experienced and well-trained individuals in that role, so it’s perhaps unfortunate that the role of CRC is one that is notorious for being relatively underpaid (compared to the tasks and responsibilities expected of them…) and for having high turnover (about twice the national average, at 30%) and COVID made this worse, with rates as high as 60% for some research sites in recent years.
The salary ranges for a CRC are as low as 37k/year, but can get up to a little over 80k. Many CRCs have medical backgrounds and they tend to be the ones paid at the higher end (for example, nurses, nurse practitioners, or even physicians who perhaps are new to the US and looking for a way to get started). Some of them though are wanting to get started on their careers, and the CRC role is really just seen as a stepping-stone. The turnover therefore isn’t because it’s a bad job (it’s actually a really fun job in my opinion!) but simply because being a CRC is not usually anybody’s career goal! People move on to the clinical monitoring role for a pharmaceutical company or CRO, or start their medical career training, or simply move up the ladder at their specific institution. The skillset and experience that is gained from working as a CRC are in demand all over the world of clinical research. The trouble with this turnover is that there are study-specific things to know, which might be due to the protocol, or the drug, or the sponsor, or the workflow at the specific institution, and losing study staff who have been trained up and who are SO crucial to the study execution means that the new CRC who picks up the mantle has to be trained all over again.
It’s very important therefore to properly mentor and support the CRCs at a site. If they are overwhelmed, mistakes are more likely. The already high rate of turnover will only be made worse, which will in turn impact the other CRCs and ongoing research at that site. I have been informed on several occasions that sites have slowed down enrollment, or even paused their research entirely, because of a staff shortage in the CRC role. It’s absolutely a worthwhile investment to have enough staff on the team to provide backup coverage and support where needed, and to have a succession plan in place for when a CRC moves on. Careful documentation of study-specific needs are crucial, as is effective and regular study team meetings with all the staff. Even if you’re not working on a particular protocol, you never know when you’ll be expected to do the work because someone is out sick, or starts their new CRO job.
As an Investigator or site manager, it would be important to bake in this salary support to things like budget negotiations with sponsors, or FTE requests and expectations with an institution, and to support ongoing training and education for the CRCs so they feel valued and can advance in their chosen career. It really is a great way to enter the area of clinical trials, whether investigator-initiated or sponsored studies and I have zero regrets from my time on the frontlines.
I could probably write a short post (maybe even a long one!) on any one of the tasks performed by a CRC in their day to day job, but for now I hope this brief insight is enough to make people realized how important they are.
A recent headline caught my eye, discussing an increase in the annual reported cases of Powassan virus – a virus which (for reasons that will become obvious) is near and dear to me. I decided to take a look at the actual data in more detail, and discuss a bit of that here, because understanding viruses like Powassan has implications beyond just this one infection.
Let’s start with what Powassan virus really is – it’s an arbovirus, meaning transmitted by arthropods (in this case, Ixodes ticks), and has an ability to cause a form of encephalitis. It is in fact closely related to the Tick Borne Encephalitis (TBE) virus, but Powassan virus was actually named after the town in Ontario where it was first discovered. It’s highly likely that many (perhaps the majority) of infections don’t go on to cause very serious disease, but the actual risk of encephalitis from it is unknown because the total number of cases isn’t really known. Of the symptomatic cases, about half are quite serious and 10% are fatal. The CDC has made it a nationally reportable infection, but the tricky part is knowing when to even think about it…
I diagnosed the first ever case of Powassan in Connecticut in 2016. It really was a bit of a crazy story, and truly a perfect storm of being in the right place at the right time. A young infant boy was admitted to the ICU with seizures and a very clear story of a few hours of a tick being attached to his leg 2 weeks earlier. The tick was brought into the house on a family member’s clothing, and probably got onto the child during a feed while being held by this person. The medical team had quite correctly ruled out most tick-borne infections due to the very short attachment time, but I knew that there was one exception – at least in animal models, Powassan virus could be transmitted in as few as 15 minutes. So it was possible, but was it probable? The clincher was the MRI report, which had a very distinct pattern showing “restricted diffusion of the basal ganglia and rostral thalami, as well as the left pulvinar”. There was no sign of more widespread of diffuse signal changes as you might see with ADEM (acute disseminated encephalomyelitis) or cerebellar changes as you might see with enterovirus. No hemorrhagic changes, as with herpes simplex. The laterality and location of the damage matched the physical symptoms (motor dysfunction affecting the right more than left), but more importantly it was also similar to previous reported scans from patients with arthropod-borne flaviviruses, including Powassan. Choi and Taylor wrote in a 2012 case report “MRI images of the patient’s central nervous system (CNS) were unique, and when such images are encountered in the clinical setting, Powassan viral infection should be considered.” We were able to test the baby’s spinal fluid at the CDC for Powassan, and it came back positive.
The points to make about this case are several – firstly, the recurring comment from most (probably all) of my colleagues when I made the diagnosis was “What made you think of it?” Honestly, it was mostly the fact that I had training in a state where Powassan was well-known and we would consider it routinely. I simply added it to the differential and saw that not only could I not rule it out, I had some evidence to support it! But the issue here is that there were probably many cases of Powassan over the years that doctors had been seeing, but simply never thought of testing. Powassan was not on a routine viral encephalitis panel in Connecticut (it’s a send-out to the CDC), whereas some other State laboratories like New York test for it automatically on their own encephalitis panel, in addition to sending/reporting to the CDC. It is very much a case of out of sight, out of mind.
Secondly, even if some physicians had considered and tested for the virus somehow, Connecticut didn’t make Powassan a reportable disease until 2019 (more than 2 years after I made the first diagnosis in the state). It’s really hard to measure something if you’re not counting it…and of course even hard to count something if you’re not looking for it!
So this makes it really, really hard to know what to do with news that “Powassan cases are increasing.” Reports are increasing, but so is awareness, and testing availability – it doesn’t mean that actual infections with the virus are going up. It is possible that they are…but the case reports alone aren’t enough to make that conclusion. You really have to understand the reporting infrastructure and testing limitations in the context of a specific disease when trying to interpret the changes you might see in any sort of incidence or prevalence data.
There isn’t a vaccine yet for Powassan virus, although when I last checked research was ongoing (and there is a vaccine for TBE). The best way to prevent infection is to avoid any kind of tick exposure at all – cover skin, use DEET, avoid tramping through the wilderness in areas where the virus is known to be in ticks, and check your pets! Also – change your clothing when you get indoors from activities that might have exposed you to ticks…
Bonus if you’ve made it this far – you can check out my TV appearance on Monsters Inside Me below!
I heard a little more recently on the contaminated blood scandal of the 1980s, where huge numbers of people with hemophilia were given treatments for their condition that were contaminated with viruses, including HIV and Hepatitis C.
Early on, there was little knowledge about the true risks of these viruses, and no proper testing available (certainly not widely available), but it turns out that some of this story is even worse than simply being given contaminated blood products.
An inquiry in the UK is currently focused on a specific series of infections that occurred at one particular school, which had a hemophilia treatment center on-site and which, it turned out, was also conducting research into novel treatments for the disease. At a superficial level the research seemed sound – it was asking the question: would a new heat-treated version of the clotting factors needed to treat hemophilia be safer than the standard treatments? Unfortunately, the heat-treatment wasn’t sufficient to completely inactivate the viruses – but the story is much worse than just that.
Several people have come forward who not only were infected with the viruses anyway, but possibly didn’t need treatment for their hemophilia at the time they were given the experimental treatments and, worse, neither they nor their parents were properly consented for the research. When the true risks of infection were obvious, subjects weren’t told, and some weren’t even informed of their infections for a year or more. One doctor involved in the study, a Dr Samual Machin (now deceased) is quoted during the inquiry:
“This would have been discussed with his mother, although I acknowledge that standards of consent in the 1980’s was quite different to what it is now,”
At the time, the subjects were all children, and several of the parents denied being properly told about the research, and that they would not have consented had they been told. Further documents showed that untreated patients were highly sought after, and that trying out the new heat-treated therapy may have been prioritized over the patients actual medical needs.
There are several core principles of research at stake here – beneficence (doing good), non-maleficence (do not do anything bad) and autonomy (being able to make your own decisions). Even a cursory look over the stories of this case show that none of those principles were upheld in this research, and of course the fact that the heat-treatment didn’t work to inactivate the viruses makes it all the worse (arguably if the research was a stunning success there wouldn’t be an inquiry into any of this…but that’s a whole other topic for discussion).
While there are regulatory rules for ensuring proper scientific conduct of research, there are also ethical rules. The two have some overlap, but they really are distinct frameworks, within which researchers have to function. The sad fact is that many of the rules and protections that we now take for granted were imposed as a direct consequence of past unethical human experimentation (which leads to another discussion about how what is considered “ethical” changes over time). From the perspective of human subjects and safety in particular, these are reflected primarily in the document we know as the ICF – the Informed Consent Form.
While it’s true that many aspects of subject safety, rights, and welfare are contained within the research protocol, the protocol itself is a highly technical description of the research and as more of a scientific justification and research plan. The ICF on the other hand is intended to be seen and understood by the study subject themselves, and includes a discussion on the research protocol (visits, procedures, timeline, risks etc.) in addition to making them aware of certain other rights afforded to them, including the right to leave the study at any time and any other treatment options available. The research site’s institutional review board (IRB) would have reviewed and edited the ICF to ensure that it accurately reflected the research risks and benefits prior to being seen by a potential subject. IRBs include subject-matter experts as well as members of the lay public to ensure clarity and understanding. Not only are potential study participants supposed to be given the opportunity to ask questions before signing the ICF, but they receive a copy and a copy remains in their medical record.
The fact that no such process occurred in these contaminated blood product studies is obvious.
Unfortunately, stories like this from research done decades ago, and which clearly doesn’t meet current standards, color everybody’s opinion of medical research. I have had parents refuse to consent for a study because “The sponsor is [insert drug company with research scandal] and I don’t trust them,” even though the drug in the study had nothing to do with the risks from the older scandal. I heard about one parent who turned down a study because of a poorly written ICF, that ironically overstated the risks in a summary paragraph that implied the drug had never been given to anyone before – some people might have continued reading, but this parent did not (they did however provide that feedback to the investigator, so we were able to modify the ICF to make it more clear). One time I had a sponsor try to avoid explaining all the risks (for the control therapy, not the study drug) in the ICF and instead provide a patient hand-out or “your doctor will explain this to you” verbiage. Despite our warning that the IRB would reject this ICF, they insisted on submitting it and of course wasted a whole IRB review cycle as they were forced to revise and resubmit the document with our recommended wording. This wasn’t an intentional thought to mislead, they genuinely thought it was a more efficient process and would avoid scaring the subject unnecessarily, but we knew that the ICF should be a stand-alone record of the subject being truly informed before consenting.
One of my experiences with obtaining consent sticks out to me – it was a slowly-recruiting antibiotic study and I was hoping to not have another screen-failure at my site. I had found a possible subject and was discussing the study with his mother and my study nurse. The mother was asking careful questions, clearly a little nervous (her son was in hospital after all!) and I was thinking that she probably wasn’t going to sign him up when she suddenly said “You know what, you’re the doctor and you know best, I’ll do whatever you say.” Massive red flag to me, as an investigator. Research isn’t the same as medical care – if “I knew what was best” we wouldn’t be having a conversation about a clinical trial! Also, if something were to happen during the study and she had rescinded the decision to me, then she as a mother would feel far worse than if she had made the decision truly believing she had made the best call. So I called “Investigator fiat” and screen-failed them. Every set of inclusion/exclusion criteria includes a line about “any other condition which, in the opinion of the Investigator. would interfere with the conduct of the study” and at that point I’m not sure that she fully understands the risks and benefits.
While we might celebrate some of these stories as holding ourselves to a high standard, the sad truth is the current standards of oversight and training that we have in medical research are much better than they were in the past mostly because of how poor they were in the past. Personally, I find it shocking that some of this occurred within my lifetime, and I think it behooves us to be mindful every day about how we conduct our research, placing the rights and welfare of our participants first.
In my ongoing saga series on clinical research, I thought I’d share my thoughts on the advantages and disadvantages of requiring regional medical monitors be located in specific countries.
One of the points raised during the planning stages of a clinical trial is often the geographic location of the team members. The Clinical team in particular pretty much has to be based locally because they will be performing site visits and interacting very closely with the staff and investigators, but it’s not unusual for other team members to be based around the globe. Data management team members for example are frequently located in Asia due to a large population of skilled talent available at more economic salaries than if they were based in Western Europe or the USA, and the fact that their work can readily be performed remotely and asynchronously with the rest of the project. But what about the Medical team?
The Medical Monitors are typically physicians with at least some experience in clinical practice (although the actual time people have spent varies considerably), and many have some degree of subspecialty training. The interesting thing is that a very large part of the job is subspeciality agnostic – the safety data we review all looks the same, and it’s largely a case of lining up the findings with the study-specific protocol to weed out the more detailed anomalies. Whether the study drug is being given for one thing or another is largely a moot point. When looking at things like protocol design and study planning and execution, there is absolutely an element of subspeciality knowledge required, but it isn’t that unusual for a physician trained in one area to find themselves working on a clinical trial in another. Furthermore, clinical trials are often global in nature and may have sites not just in different time-zones, but on different continents, and the Medical Monitoring work itself is readily performed remotely. For this and other reasons, the physical location of the Medical Monitor may not be considered relevant, but I would argue that there are very good reasons to keep the Medical team regional and optimized for the study, beyond the simple consideration of time-zones.
In the early planning stages, similar to being a subject matter expert from the disease/treatment perspective, it can be critical to have the insight of a local physician when considering things like screening failure rates, ease of enrollment, site and investigator selection, and even country-specific nuances on treatment guidelines or standards of care. For example, if a protocol requires that a specific comparator drug be used, but that drug isn’t approved or recommended in some countries, then it would be a mistake to try to conduct the study in those countries. Certain medical conditions are more or less common around the world, whether due to genetic or environmental factors, and these can dramatically impact enrollment rates and timelines. While it is true that a lot of this information can be obtained and understood by any physician, I think it’s still prudent to consult someone with that knowledge from a medical perspective.
During one large project I was on, we received feedback that a particular medical test was likely under-budgeted, which might lead to sites refusing the take the study on if it were not reimbursed properly. It was something that was highly unusual in routine medical care, but which I happened to have knowledge of and an awareness of the complexity of the procedure. Having an awareness also of the way the American healthcare system tracked and billed for these tests, I was able to direct the budget analysts to the correct amount to cover which impacted not just the proposal I was currently working on, but any others that used the same test. Someone without that knowledge of how the system worked might not have even known that there was an answer out there to find, never mind find it.
One area that is also important, although it’s often not realized until it’s needed, is clear communication with the sites and Investigators. While it’s true that English is widely used and understood, and tends to be the “lingua franca” of the research world, it’s also true that English is a second language for most. It seems prudent, if not simply polite, to be able to converse with site staff and investigators in their native language, and furthermore to have the social cues and etiquette familiar to them. I can speak French fairly well, but I know I cannot communicate the nuances in French that I can in English. At best, this can slow down communication if a third-party is required on a phone call to interpret in real time – at worst, it can lead to miscommunication and inadvertent offense. I have found that the issues are compounded if a person who natively speaks one language converses in English with someone who natively speaks another, and because clarity and attention to detail are SO important, I think it does behoove us to consider this aspect of study execution when assigning Medical Monitors. I don’t think this necessarily requires physicians geographically local to the sites, but doing so definitely decreases the odds of miscommunication.
Having already said that geographic location is irrelevant when considering the basic data and safety review tasks (with the exception of nuances in country/region specific regulatory reporting of urgent cases), there is other reasons for appointing Medical Monitors in alternative locations to the main study sites, especially if the time-zones at least line up. For one, it may be economically advantageous to hire on physicians from certain countries, or the volume of work may simply require multiple people with the team members scattered. There is also the very real possibility that the most experienced or suitable physician for that study isn’t local, or the sponsor may have a specific preference based on past experience with that person, so obviously a number of factors need to be considered.
Importantly, because the needs are different for project development versus execution, It may be the case that the subject-matter expert assigned to the initial proposal team isn’t assigned as the Medical Monitor. In that situation, it’s crucial for a comprehensive hand-off/kick-off meeting to occur to make sure that any issues or risks identified during the initial work are communicated over to the main team.
From the perspective of the sponsor, I would consider all of these issues when looking at the Medical Monitor assigned to your study – what aspects are most important to you, and what tasks are they most likely to be performing? I would say that if site communication and insights are important, then advocating for a local assignment would be a good idea. If you consider that subject-matter or clinical trial design expertise is more important, then you may want to be targeted in your selection regardless of geographic location. I am also a huge advocate for remote working for Medical Monitors, especially when considering all the factors above: there is a finite pool of experienced experts and if you happen to find someone with the right mix of clinical and research time, covering the right subject-matter, and with the personality and work-ethic to excel in clinical research, you absolutely should not limit your recruitment to physicians who are willing to relocate. Most physicians who are at the stage in their careers where a move to industry is feasible are in their middle years, settled with children and houses, and in many cases burned out with the time and demands of clinical care. Being told they have to uproot their family and commute through traffic to a desk job in an office is not appealing at all. I have seen several companies, both in the CRO world and in big Pharma (less so in Biotech interestingly enough), require their Medical Monitors to relocate and work on-site. I simply do not think there is a need for that in this line of work, especially in the post-pandemic era where work-from-home infrastructure is so well developed. If you’re requiring your talent to relocate you’re effectively giving the talent away to your competitors…
What’s been your experiences working as, or with, a regional medical monitor? I’d love to hear about the advantages and challenges you’ve experienced.
In my first post on Clinical Research, I mentioned how important it was for Investigators to adhere to the protocol, but I didn’t really go into detail on the whys and wherefores. Protocol Deviations (PDs) are often misunderstood, even by those who are tasked with identifying and categorizing them, so I wanted to write this breakdown of the concept and why it matters to everybody involved in clinical research.
By definition, a PD is anything that causes one or more subjects to deviate from the strictly defined procedures and events laid out in the protocol. You might imagine then that it wouldn’t be difficult for this to happen – for example, if a subject gets sick and cannot attend a study visit when they’re supposed to, that would be a PD. That wouldn’t be anybody’s fault, it’s simply something that couldn’t be avoided, but it is still a PD. More seriously if a pharmacy fridge fails without anyone noticing, and a subject is given a medication that might be ineffective, that would also be a PD.
There used to be a term of Protocol Violations (which were deemed more serious) and PDs, but that guidance has since been clarified/rescinded and now all should simply be reported as PDs, and further categorized into Important or Not Important. Important PDs are those that would impact the rights, welfare, or safety of subjects, or would potentially negatively impact the integrity of the data. Non-Important PDs are divergences from the protocol that do not significantly impact subject safety or data integrity. In part, I think this was to reduce the perception that PDs were a punitive act against the sites and Investigators. Investigators often feel that they are being blamed for events that are outside of their control (for example, sick subjects not showing up), or equipment that doesn’t work. There is a fear, which is in part founded on the reality that they are wholly responsible for the research conducted, that too many PDs will lead to their ability and integrity being called into question. It is true that in the very worst cases an IRB can revoke an Investigator’s ability to conduct scientific research, but in more than 99.99% of instances I would say that the PDs are meant as an administrative and a statistical tool, and certainly are never intended as punitive.
The true purposes of detecting PDs fall into two broad categories – regulatory, and operational. From a regulatory perspective, it is absolutely crucial to detect and identify a “per protocol population” – the group of subjects who, as best as reasonably possible, followed the protocol as agreed to by the regulatory body (e.g. the FDA in the USA). Without this, the sponsor for the study cannot possibly go back to the FDA and say “Look, we proved our drug is safe and effective – please approve it for us!”. If too few subjects followed the protocol, as agreed, then the FDA will simply refuse and tell the sponsor to go back and get more subjects. This is why it is so crucial that Investigators do not willingly deviate from the protocol, even if (and I cannot stress this enough) they would do something different under the normal standards of medical care. Losing one subject here and there is not too great a problem, but losing hundreds (or even thousands) of subjects due to PDs can be devastating to a clinical trial. For this reason some studies retain the “violation” distinction for the most serious PDs that would drop a subject from the per-protocol population, whereas others might opt for a designation of Major or Minor PDs. The distinctions are crucial, and must be clearly defined by the entire project team, including the Clinical Operations, Medical Monitoring, Data Management and Statistical team members. Many of the PDs will be detected by Clin Ops during their site visits or through remote data monitoring; a large chunk are detected during Medical data review (for example the use of prohibited medications, or potential exclusion criteria in medical history); and without the input of a statistician and data manager it isn’t possible to properly assess the impact of these PDs on the final analysis of the clinical trial. In some cases, sites may be asked to track additional information, or the data management team may have to code custom programming to identify and track certain PDs. In one study I was on, the out-of-window visits were subdivided into Major or not depending on how far out of window they were, and only for certain key study endpoints. This is the kind of nuance that can’t be simply tracked as an “out of window” visit – it needs to be “out of window for Visit X by Y days”, and would absolutely need custom programming (and very clearly written protocol deviation guidance) to detect, if the sponsor wanted to go down that route.
I was involved in a company-wide initiative to update and improve our Protocol Deviation process, to optimize the process and understanding of each team member on their role. Medical Monitors in particular have to weigh in on Important PDs, which are defined as any that might impact subject safety or data integrity – ultimately we have to decide if that subject should be dropped from the study or if they may continue. Sponsors often don’t appreciate the distinctions between the different levels of PD, and the work they entail, so it’s important to communicate that clearly during study start-up. PDs are also very much study-specific – for example an “expedited PD” may require the Medical Monitor to contact the site and intervene on an urgent basis, but it makes no sense to mark certain PDs as expedited in a single-dose study (such as a vaccine study) when the PD would be detected long after the subject was dosed. It also makes no sense to mark PDs that would be detected by the Medical Monitor as “expedited”, because they already know about them! If a missed study visit occurs then it’s reasonable to track that as a single PD, and not necessary to track separate PDs for every missing procedure during that visit (yes, I’ve seen this done inadvertently!) The goal of this process improvement was to streamline and align the PD process between all the relevant team members, and ultimate save time and money throughout the entire study timeline.
The second reason for detecting PDs is operationally – the Clin Ops team monitors and tracks PDs, and especially important is the categorizing of PDs. For example, if they find that there are many PDs for inclusion/exclusion criteria, it might be that the protocol is unclear and needs revision. If they find that one site has many PDs compared to their peers it might signal an issue that requires site staff retraining – and the flip side of course is that if a site has very few or no PDs that they aren’t being truthful in their data reporting… Ideally these are reporting back to the Sponsors regularly throughout the study, although I have come across some smaller biotech companies who have asked merely for updates perhaps one time during the study, and once at the end. In my opinion this is a recipe for disaster – it means that the operational benefits of the PDs are diluted, if not lost, and it also means that potential revisions to the protocol that could improve the quality of the data (or subject safety) are not made. The very best companies use PDs as “constructive criticism” to optimize their study sites and protocol. PDs can also be an absolute bear to properly collect and categorize, and the worst thing that could happen to a study team is to discover right at the very end of a study that there are hundreds (or thousands) of PDs to address when there are looming deadlines of database locks.
There are two further things I want to highlight about PDs. There is a hierarchy that actually stretches very high up – PDs can be at the study level, the country level, the site level, or the subject level. Study and country-level PDs are extremely rare. Site level PDs do occur, but they are often misidentified, and that can cause a lot of work. By definition, a site-level PD affects all subjects at that site. This is a statistical definition, not a common-sense definition. For example, a broken pharmacy fridge might be incorrectly identified and recorded as a single site level PD (failure to adequately maintain and monitor study equipment) – but the problem then arises in the final analysis, where EVERY subject from that site would be tagged with that PD! In truth, the clinical monitor who visits the site has to correctly identify the subject(s) impacted by the event, which usually means tracking specific dates, subject timelines, and drug deliveries to figure it all out. They would have to identify and report PDs on the affected subjects, and ONLY the affected subjects. This might mean reporting 23 separate PDs (one for each of 23 affected subjects) instead of one site level PD, unless the site has only enrolled 23 subjects! But trust me I say this is the correct way to do it – this is exactly how the FDA will ask the events to be captured if they were to visit the site for an audit. I perhaps misspoke above when I said that the worst thing that can happen is for a last minute deluge of PDs just prior to database lock – imagine a deluge of PDs after database lock because of an FDA audit findings…
And finally, the reason for the meme I made above (which I have actually presented in Investigator training slides…). Sites often ask for “exemptions” or “permission” for a PD ahead of time, for example if they know that a particular subject is going to be out of town and miss a study visit, or if a subject has a prohibited medical condition but would otherwise qualify for inclusion in the study. The thought is, if they ask ahead of time they won’t be “dinged” with a PD.
It is a very, very, very bad idea to grant exemptions to PDs. As I said above, PDs are there to ensure data integrity and that the per-protocol population is sufficient to be submitted for regulatory approval. If a significant PD is granted ahead of time, the entire clinical trial is put at risk. I was trained to never, ever, grant a PD exemption. We can acknowledge that a deviation has (or will) occur, but it will be reported, and it will be categorized, and if need be the local IRB will also have to hear about it. Now, there are some sponsors that will grant PD exemptions on a case-by-case basis but that is their risk to bear. Not every event reported as a PD is actually a deviation when fully reviewed, and it isn’t unreasonable to void a PD that shouldn’t be there, but they would have to defend that decision should a regulatory authority question it, and if you don’t think that a regulatory authority will detect discrepancies in individual protocol deviations and subject data, then you haven’t witnessed a regulatory audit… What the site/Investigator really needs to ask is “Can this subject continue in the study, should this PD occur?” That’s an entirely different question, and is probably what the site is really wanting to know, but even if the answer is “yes” then every effort needs to be made to follow the protocol – the Investigator literally signed a legal document saying they would do this (I talked about Investigator responsibilities last week…)
To summarize – PDs are inevitable, even if they should be avoided as much as possible, and they are very important tools used during the conduct and in the final analysis of a clinical trial. Clear PD guidance should be established very early on in the study start-up to identify areas of risk and mitigation steps, and PDs should be routinely reviewed with the sponsor so that course corrections or staff retraining can be done as needed. Proper categorizing and coding of PDs (Important, Non-Important, Major, Minor, Expedited…) will dramatically improve the quality of the data and project workflow for everybody involved. Sites should not fear PDs, and they absolutely should not attempt to cover them up, but please don’t ask for exemptions.
Drop me a line, or follow this blog, if you want to learn more about PDs and how best to avoid or manage them.
I chose this rather banal title firstly because it is descriptive, but secondly it draws attention to the seriousness of what it means to be an Investigator on a clinical trial. Following up from my first post on this topic, here I’m going to go into more detail on the role and responsibilities.
21 CFR 312.60 – An investigator is responsible for ensuring that an investigation is conducted according to the signed investigator statement, the investigational plan, and applicable regulations; for protecting the rights, safety, and welfare of subjects under the investigator’s care; and for the control of drugs under investigation. An investigator shall obtain the informed consent of each human subject to whom the drug is administered, in accordance with part 50 of this chapter. Additional specific responsibilities of clinical investigators are set forth in this part and in parts 50 and 56 of this chapter.
My first experiences as an Investigator actually stretch back to my earliest days in clinical research in 2004. I was in a kind of twilight zone for a little while, technically hired on as a study coordinator but, with my medical and PhD degrees, I was actually named and delegated responsibilities as a Sub-Investigator on several clinical trials. The training that we all have to do to undertake human subject research makes it very clear how serious and important the role of the Principal Investigator is. The preamble statement from the USA Federal law must be fully comprehended. “An Investigator is responsible for…” Do you really know what that means? If a study coordinator accesses medical records improperly for subject screening, the Investigator is responsible. If a nurse draws the wrong number of blood tests for a specific study visit, the Investigator is responsible. If a pharmacist dispenses the wrong dose of study drug for a subject, the Investigator is responsible. Investigators can (in fact, they must) delegate tasks to study site personnel who are properly trained and qualified to perform those tasks, but delegation does not absolve them of responsibility. This is a significant increase even from the supervisory role that many physicians are in, whether it’s teaching residents or fellows, or supervising Nurse Practitioners or Physician Assistants. As a Principal Investigator the Physician is held responsible even for tasks that they themselves might be incapable of doing (for example, study drug dispensing, or certain types of clinical testing).
This requires a seriousness of mind when a physician decides to undertake clinical research, and in addition it requires considerable support. Whenever I am asked about whether a physician should become an Investigator, my very first thought is “do they have enough support staff?” Having been a study coordinator, and having worked with study coordinators of my own, I have to say that regardless of how good a researcher might think they are, they will absolutely fail in running a clinical trial unless they have a good (if not great) study team. The administrative tasks associated with study start-up alone are Herculean – reviewing the Clinical Trial Agreement, Protocol, and Investigator’s Brochure, adapting the informed consent form (and in some cases, the protocol itself!) to local standards for IRB submission, completing the IRB submission (and any other review boards required), negotiating a budget, organizing sub-Investigators and collaborators, and ensuring adequate space and equipment exists to see subjects, perform procedures, and collect all the required specimens. All of that is before you even see a single subject. If you are able to get all that done, then a representative or two from the sponsor or contract research organization will visit to activate the site. A site initiation visit (SIV) is generally several hours of time for training, inspections, and administrative paperwork such as the delegation log, training logs, and FDA1572 “Statement of Investigator” form. Most physician Investigators are hard-pressed to even make it to an hour of the SIV – never mind attend the entire visit. I was fortunate to have excellent study staff to work with, and in fact a dedicated Clinical Trials Unit (with their own clinic space and lab) existed to help Investigators throughout the institution. My coordinators included a nurse, and I had a lab tech and study pharmacist team to use too. I was very well supported!
Aside from needing the appropriate mindset to conduct research seriously, and a solid team, there are several areas where Investigators can let their site down, even with the best intentions to heart. Perhaps the most frequent way in which Investigators can get into trouble is agreeing to perform a study that they cannot properly execute. When an investigator is initially approached for a study, they should perform a genuine, reality-based assessment of the feasibility of the protocol. If the blood tests require a -80C freezer, and they don’t have a -80C freezer, then they can’t agree to do the study until they buy one. If the study requires that subjects stay for 8 hours getting pharmacokinetic blood tests done, but their clinical trial unit closes at 4pm, then they can’t agree to do the study. If they only see 1 patient with that specific diagnosis every few months, then they shouldn’t commit to enroll 10 patients in a year. That last issue occurs again and again…I think in a (very) misguided approach to convince the sponsor to allow them to sign-up because they’ll be a “good site”. In truth this is a bad idea, for several reasons. Firstly, site budgets are often written based on the expectation that a certain number of subjects will enroll, and some overhead costs are averaged out over the entire patient mix. Obviously the specifics vary by institution and study budget, but I have certainly heard of sites getting into financial trouble because the investigators would consistently over-promise and under-deliver. Secondly, it makes the investigator look bad – because as much as they can promise the moon, when reality hits they can’t hide it. The CRO and Clin Ops teams will be pushing hard for enrollment, because they in turn are getting pushed by the sponsor for enrollment, and you can bet your bottom dollar that the sponsor is pushing because their Board of Directors and shareholders are pushing for enrollment! Do. Not. Over-promise. Thirdly, and this is a fun little factoid – but the average and typical enrollment data is publicly available. We can tell if a site looks like they’re over-promising, because their enrollment projections will be vastly different from those seen elsewhere in similar studies. I have absolutely seen research sites and Investigators removed from potential study site lists because they were not thought to be realistic.
Investigators can also negatively impact a study by managing their subjects too much like a treating physician. I have heard over and over again something like “Oh, yeah we identified 3 eligible subjects, but they’re not due back in clinic for 3 months.” What? No! So in 3 months you might talk to them about the study, you might give them the consent to review, and maybe they’ll bring it back and sign it in another 3 months? Talk to them over the phone, now, tell them there is a study they might be eligible for – give them any websites or other information (IRB-approved, of course), and you can even email them the consent to read over ahead of time. Bring them in EARLY for a screening visit, separate from their next official medical appointment, go over the consent in person and answer any questions, and if you’re lucky they’ll sign up next week! There is a sense of urgency in clinical trials that simply doesn’t exist in routine medical care. Do not wait to discuss research with your potentially eligible patients and families.
After enrollment, do not manage clinical trial visits and procedures as you would medical care – blood tests might be timed down to the nearest minute, study visits often have allowable windows of only a few days early or late (big tip to schedule study visits early in the window, in case anything happens and you have to delay). You have to be very well organized, and again this goes back to a diligent study staff team. For one clinical trial I was a coordinator on, I created a spreadsheet for all 191 subjects with built-in math to figure out all the required telephone calls and visits that had to be scheduled. A lot of studies these days have similar tools built into the electronic data capture systems to assist site staff with this, but I started back in the day of paper records for clinical trials… In any case, find a system that works and is reliable, and use it! The risks of a lack of diligence and organization are, at the least, protocol deviations and poor data. At the worst, the site can be shut down and the Investigator may be prohibited from conducting research. I have personally seen sites that were put on hold due to serious breaches in Good Clinical Practice (GCP – the rules regarding subject safety and clinical trial execution) that placed subjects at potential risk of harm, all due to a lack of diligence from the Investigator (remember “Clinical” in this context refers to medical research and clinical trials, not standard medical care).
Every Investigator should have undertaken training in the principles of GCP, in fact it is one thing that is repeated for every clinical trial training regardless of whether or not an Investigator has done it before! This training is not entertaining – it is snooze-worthy – but it is very important. The physicians who don’t pay attention to it are the very same ones who are immediately asking for permission for protocol deviations or to skip certain study procedures or visits “because they’re not needed”. If it’s in the protocol, it’s needed. I absolutely urge Investigators, whether would-be, newbie, or seasoned veterans, to pay attention when it is being done. Clinical research certainly isn’t something that should be done in a haphazard or half-hearted way. There is also no room for ego – Investigators and other study staff will make mistakes, Protocol Deviations will occur, misunderstandings will happen. The extremely high standards and rigorous oversight that are required for clinical trials is something that most physicians aren’t used to in their day-to-day practice of medicine, and it can often be misinterpreted as a personal attack or insult to their intelligence. The fact is, we’re really all on the same page, trying to get the best possible data, and keep the subjects as safe as possible.
On that note, the Investigators are responsible for the safety of their subjects. In that regard the opinions and actions of the Investigator matter a great deal. If an Investigator deems that a subject should be withdrawn from a study, they can do that. If an Investigator grades a particular adverse event as mild, moderate, or severe, then we generally rely on their assessment as the clinician who saw the subject (there are exceptions for specific laboratory results or protocol-defined criteria). Investigators assess whether an adverse event is related to the study drug or not. While the Medical Monitor and sponsor can (and often do) request clarification or confirmation of a specific finding, and sometimes even disagree with the Investigator, the Investigator’s assessments are still reported and are incredibly important. Some types of adverse event require very rapid (7 day) reporting to regulatory agencies, and that all hinges on the Investigator’s assessments. It is not unusual for Investigators to reach out to Medical Monitors for clarification or advice, but the Medical Monitors cannot provide patient care and cannot really direct an Investigator to do one thing or another. We can provide information and clarity on the protocol, insight into what the sponsor might think on a particular decision, and discuss any potential protocol deviations that might occur, but ultimately it is the Investigator that makes the final call. I have completed many telephone conversations with the line “I support your decision”.
In that regard I will discuss one final thing that Investigators do that causes issues, albeit rarely – subject unblinding. In randomized clinical trials, the subjects and often Investigators (even the Clin Ops teams and Medical Monitors) are all blinded and unaware of treatment assignment. This is so that no-one contaminates or biases the data from assumptions made of the treatment being given. However, it also happens that subjects occasionally get adverse events that may or may not be study-drug related. There is generally a clause in the protocol that allows for emergency unblinding under certain specific circumstances. That last bit is crucial – because unless it is genuinely thought that (a) the event is linked to the study drug and (b) knowing the treatment assignment will affect the medical management of the subject’s adverse event, there is no reason to unblind the subject! If on month 4 of a vaccine study the subject has a severe stroke, what would you possibly do differently with them, knowing what vaccine they got 4 months prior? Would you use a different brand of tPA? Would you shorten their rehab knowing that they “only” got placebo? No, it wouldn’t matter one iota. Trust me – the subject will be unblinded as part of the final analysis – and often studies have unblinded safety monitoring teams who will look at these type of events separately from everybody else on the study (I have sat on both blinded and unblinded safety monitoring committees for various clinical trials). There is very rarely any rush to unblind as an emergency, and if that is done it actually risks contaminating the data and losing that subject from the final analysis. By all means use emergency unblinding when necessary, but think clearly on whether or not it would actually change the management of the subject or whether it would “just be nice to know”. Do not risk the entire drug approval process just to appease your personal curiosity. It is perfectly reasonable to have a discussion with the Medical Monitor and the sponsor as to whether or not an unblinding should occur, and that can wait until the dust has settled and more information is at hand. Remember, an Investigator can withdraw a subject from a study at any time, and there are allowable reasons to stop study drug while still continuing in the study for safety follow-up. Neither of those options requires unblinding.
Those are highlights of the much broader and more nuanced role of a clinical trial Investigator than I can cover in a single blog post. What I hope I have imparted though, is a sense of how seriously the role should be taken. Investigators are literally the beating heart of clinical research, hugely important, with a great deal of trust given to them, and I found the role to be an incredibly rewarding aspect of medicine, both personally and professionally. I’ll cover how you actually become an Investigator in another post.
It is a common misconception among physicians that engaging in medical research (or clinical trials) is not much different from practicing medicine – I’ve seen the question may times along the lines of:
“I’ve been asked to be an investigator for a clinical trial. How much work is it beyond finding patients, and how much will I get paid?”
There are so many aspects to this area that I’m actually going to devote several separate posts to this. I’ve thought for a long time that it would be a great idea to draft some educational material to help sites, subjects, and study staff to navigate the complexities that are clinical trials, and here we are.
I have been involved in clinical research for about 20 years now – covering everything from being a study coordinator recruiting subjects, collecting blood specimens, and completing case report forms, all the way through to being a medical monitor responsible for the safety of literally thousands of subjects, and ensuring that the data collected is as good as we can get. I have seen drugs fail, companies fall, and I’ve also seen new approvals get to market. It’s an incredibly rewarding career.
The art of medicine though requires a high level of insight and a degree of imagination – I have used a phrase along the lines of “Rules are for the protection of the weak and the guidance of the wise.” – usually right before I break a rule of some kind 😁. Physicians are in fact expected to think outside of the box and imagine what might be the diagnosis, and they have access to an ever-expanding arsenal of medical treatments to choose from for their patients (thanks to clinical trials – hint hint). Documentation is proscribed and expected, but the specific language used has considerable leeway. As strict as the framework of practicing medicine is, it’s still not that bad when you think about it.
Clinical Trials are a whole other beast. When an investigator agrees to oversee a clinical trial, they actually have to sign a legal document agreeing to follow a specific protocol for that study. This protocol isn’t just a plan to conduct the research – it is a highly detailed and specific document, with many ancillary documents, detailing every step of the subject’s journey from screening until closeout, and what might happen along the way. It is a document vetted by statisticians, clinical operations (people who work with the sites to execute the protocol), data managers, regulatory and legal experts, and independent physicians (protocol review is a routine part of a medical monitor’s job). The protocol has to be signed off by the FDA or other regulatory body at the country-level, so they agree that it doesn’t put subjects at undue risk and has all the required steps to meet the study objectives. As a simple example, enrolling too many subjects might put people at unnecessary risk, whereas enrolling too few subjects might make it statistically impossible to show that the clinical trial has succeeded. At the local level, every site has a Institutional Review Board (and sometimes a Scientific Review Board as well), and they also have to review and sign-off on the research as being appropriate. Everything that a subject might potentially see has to be vetted to ensure it is easy to understand, fully explains the risks and benefits of consenting to the research, and isn’t coercive. Every member of the study team has to have evidence of the proper training and qualifications to conduct not just research, but this protocol specifically.
Clinical research is so tightly regulated that an entire section of international (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guideline for Good Clinical Practice (GCP) ) and United States law (various parts of 21 CFR) is devoted to its conduct. The point being, that when an investigator agrees to conduct the study according to the approved protocol they cannot change it. They don’t get to chose when, or if, a subject shows up for a visit, they don’t get to chose the treatment (unless that is defined in the protocol), they don’t get to decide which tests are or are not performed – not unless there are very clear reasons and exceptions laid out. One obvious reason is for immediate subject safety needs, but that is exceedingly rare. I’ll talk more about Protocol Deviations and what they mean in a later post…
From the subject’s point of view though, I think it is crucial to appreciate that clinical research is by its very nature, and due to the scientific and and legal constraints placed upon it, incredibly strict and well thought-out. Clinical research is not a case of “let’s try this and see if it works”. I do think that subjects are aware of the second point about clinical trials – that they often provide access to new and as-yet unavailable treatment options. I know that the investigators are very mindful of this fact, and in truth one key motivator for being an investigator is in making these treatments available to their patients right away, and in contributing to the greater good by hopefully bringing a new treatment to market to make it available to all.
From the Investigator’s point of view, they should not undertake clinical research unless they are prepared to be held to an incredibly high standard – far higher than they are used to in the day-to-day practice of medicine. That is the price to pay for getting access to brand new and cutting-edge treatments. We’ll go over the specifics of the investigator’s role and responsibilities in a later post, but suffice to say – don’t do it for the money 😉.
Many of my friends live in the UK, and have now been put under a “Tier 4” lockdown in response to rising case numbers, particularly in London and the South-East of England. Along with this news is the reporting of a “new strain” of the SARS-CoV2 virus thought to be responsible for this surge. The Interwebs are now aflame with doom and gloom scenarios of vaccine futility, global spread, and the basic fear that new = bad. Now, this is still 2020 so of course people can be forgiven for feeling that way, but if anything the facts are reassuring.
The simple truth is, the UK would have to enact harder lockdown efforts based purely on the rise in cases recently, regardless of whether or not a new strain was identified genetically. It is a total misrepresentation to suggest that the UK is locking down BECAUSE of a new strain – they are locking down because of a rise in cases. New strains have been reported throughout the pandemic, most notably the 614G mutation that arose in the spring but is now the predominant strain worldwide. In many ways the panic surrounding THIS particular strain is due to awareness rather than any hard data.
UK COVID case numbers from Worldometer, as of Dec 20th
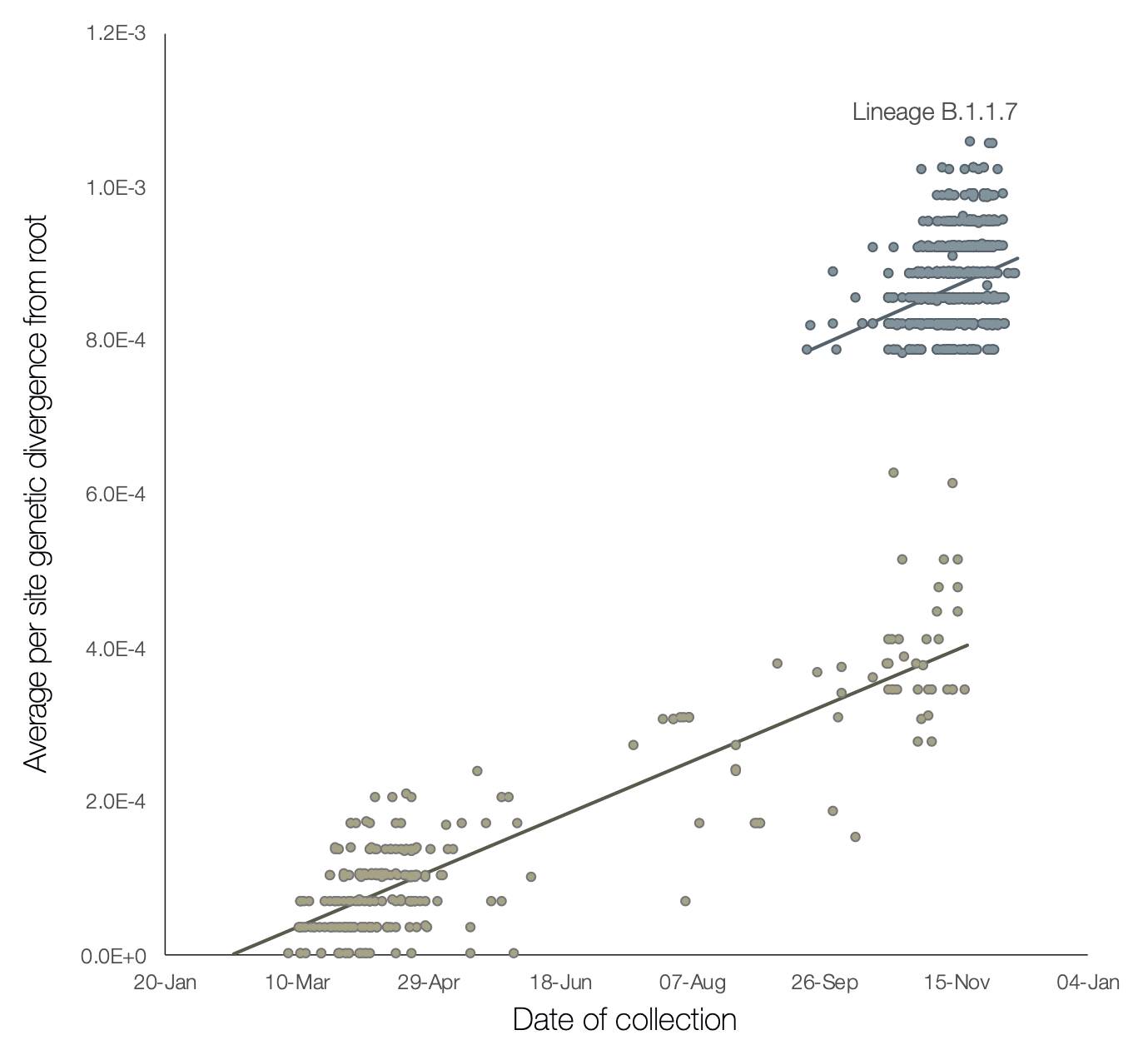
There is no doubt that the UK has failed at containing COVID-19, but not only is there a second wave of infections starting from September, there is a sharp 3rd spike over the last few weeks. This is truly an unusual graph, and not one that I’ve seen quite so dramatically elsewhere. A report in the British Medical Journal, and further outlined elsewhere tells more of the story, and perhaps explains that new surge in cases. A graph from that last paper by Rambaut et al shows the frequency of detection over time, and the genetic divergence, of SARS-CoV2 isolates in the UK.
Firstly, if you look at the lower cluster, it shows a slow increase in genetic diversity over time, as expected with this virus anyway (1-2 mutations per month). You can’t really compare disease incidence with this graph because only a subset of isolates are going to get sequenced, but you still get the sense that there was a peak in the spring and less over the summer. The cluster in the top-right is entirely distinct – firstly the diversity is way off from the trendline of the previous infections (which are still ongoing) but also the proportion of current isolates coming from this new cluster seems to be high.
The obvious worry is that the new strain is more infectious – but the evidence for that is still circumstantial. Just because more cases are occurring doesn’t mean the new strain is responsible – after all, it also true that more widespread infection will make the appearance of significant mutations more likely. There are several mutations identified in this strain within the spike protein that could, in theory, render it more infectious by altering it’s receptor-binding properties (after all, that’s thought to be why SARS-CoV2 is more infectious that the original SARS). The affected sites DO appear to alter receptor-binding and entry locations, and so there is a little more of a “smoking gun” regarding infectivity than merely seeing high numbers of cases – see the section “Potential biological significance of mutations” in the Rambaut paper.
Importantly, there is apparently not sufficient change in the S protein to affect the likely vaccination targets…although honestly we will have to see how the real world data looks with that. The Pfizer vaccine is a whole-protein target with many more places for the immune response to target than just the receptor-binding area at the top. Worries that this “new strain” will render the entire vaccination effort meaningless are not founded in any fact at this point, since all this virus really is, is a slightly different strain of the same virus, to which it remains about 99% the same (8 mutations in a protein that is over 1200 amino acids long…)
And lastly, there is the intriguing finding that some of this new lineage of virus contains a series of mutations in a gene called “ORF8” (simply, open-reading frame 8). This gene is thought to be involved in hiding from the host immune response, by downregulating the protective antiviral system called MHC-1. Similar mutations have been reported before, and not surprisingly the virus is less dangerous. In a small series from Singapore, 28% of patients with wild-type COVID required oxygen, compared to 0% of those with the ORF8 deletion. Rambaut et al hypothesize that this UK strain arose from prolonged infection in an immune-compromised host, perhaps having been treated with multiple antiviral therapies. This would explain the cluster of mutations all appearing together, as well as the potential loss of ORF8 (since it would pose no advantage to the virus to keep it).
And let’s have a quick look at the daily deaths from the UK. While the daily cases are 5-times higher than back in the spring (25,000/day versus 5000/day), the daily deaths are half as high (500/day versus 1000/day). While this reflects a mix of improved management of cases, perhaps different age demographics, it also begs the question as to whether the virus itself is simply less dangerous than it was before.
Looking at this information, I see it that while this may indeed be a new strain, it is not a reason to panic. We have no real reason to suspect that it’s going to affect the vaccine efforts, and absolutely no reason to suspect that it’s any more dangerous than before. In fact, there is a suggestion that it may be LESS dangerous than earlier circulating strains. We should be thankful that scientists are tracking and reporting this data, and remember that any new mutations that do render the current vaccine ineffective can be engineered into a new mRNA vaccine extremely quickly. The important thing is that in order to contain this virus we do exactly what we’ve been doing already – isolating, masking, and continuing our vaccine program.